Our lab studies data science problems in genomics, combining bioinformatics, statistics, and machine learning. We develop computational tools for analyzing “omics” data, with a current focus on single-cell transcriptomics (gene expression within individual cells) and spatial transcriptomics (gene expression across spatial locations). One central goal is to better understand cellular heterogeneity and distinguish biological signals from technical variation. Several ongoing research directions include:
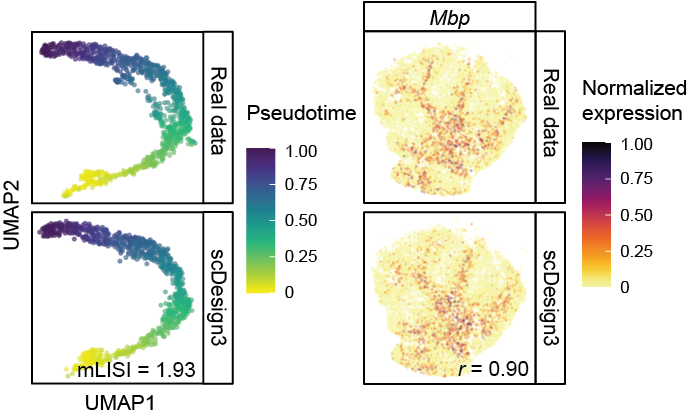
Omics data simulation
How can we generate realistic omics data computationally? The short answer is building a model that learns from real data. Realistic simulation of single-cell and spatial multi-omics data plays a critical role in both evaluating the performance of computational tools and exploring experimental designs. We developed scDesign3 (Nature Biotechnology, 2024), an “all-in-one” multimodal single-cell and spatial omics simulator based on a parametric model. We also contributed to scReadSim (Nature Communications, 2023), a single-cell RNA-seq and ATAC-seq sequencing read simulator, and scDesign2 (Genome Biology, 2021), the predecessor of scDesign3.
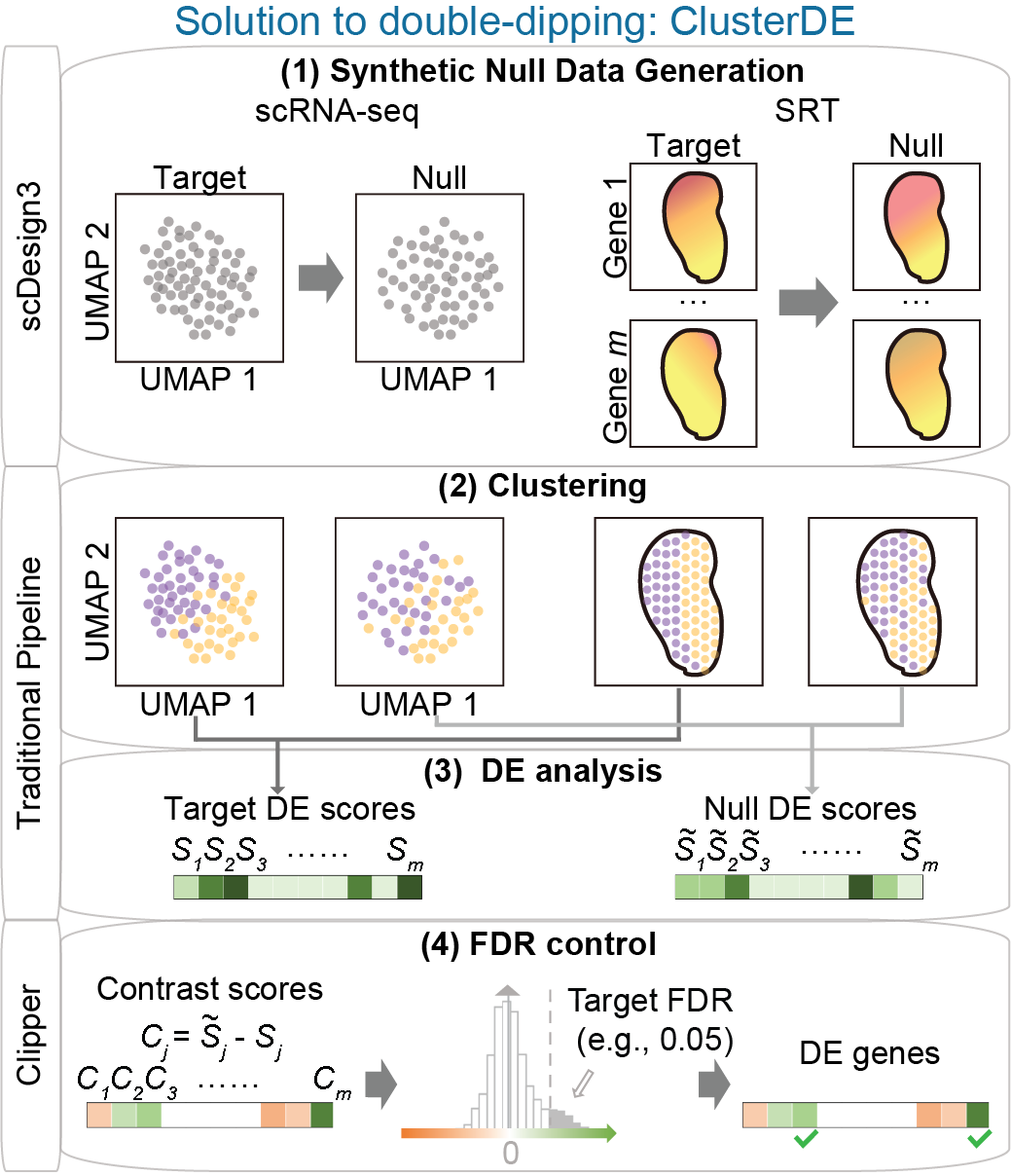
Statistical testing of gene expression and co-expression
How can we accurately detect changes in gene expression levels and co-expression strength? We need to account for different sources of variation and randomness to draw reliable conclusions. For example, in scRNA-seq, cell types are often annotated by clustering; differential expression (DE) tests between clusters can then identify many false DE genes because of double dipping. We developed ClusterDE (accepted by RECOMB 2025), a post-clustering DE test method that controls false discovery rate (FDR) under double dipping. Another example is trajectory, or pseudotime, inference, where analysts often ignore the fact that pseudotime is random. We developed PseudotimeDE (Genome Biology, 2021), a DE test for gene changes along cell pseudotime that accounts for pseudotime uncertainty. In addition, we developed scGTM (Bioinformatics, 2022), an interpretable statistical model for capturing a gene’s expression trend. Recently, we developed spCorr (Genome Research, 2026), a co-expression modeling method for detecting dynamic co-expression changes across locations in spatial transcriptomics. By using a flexible regression model, spCorr can account for multiple confounding effects.
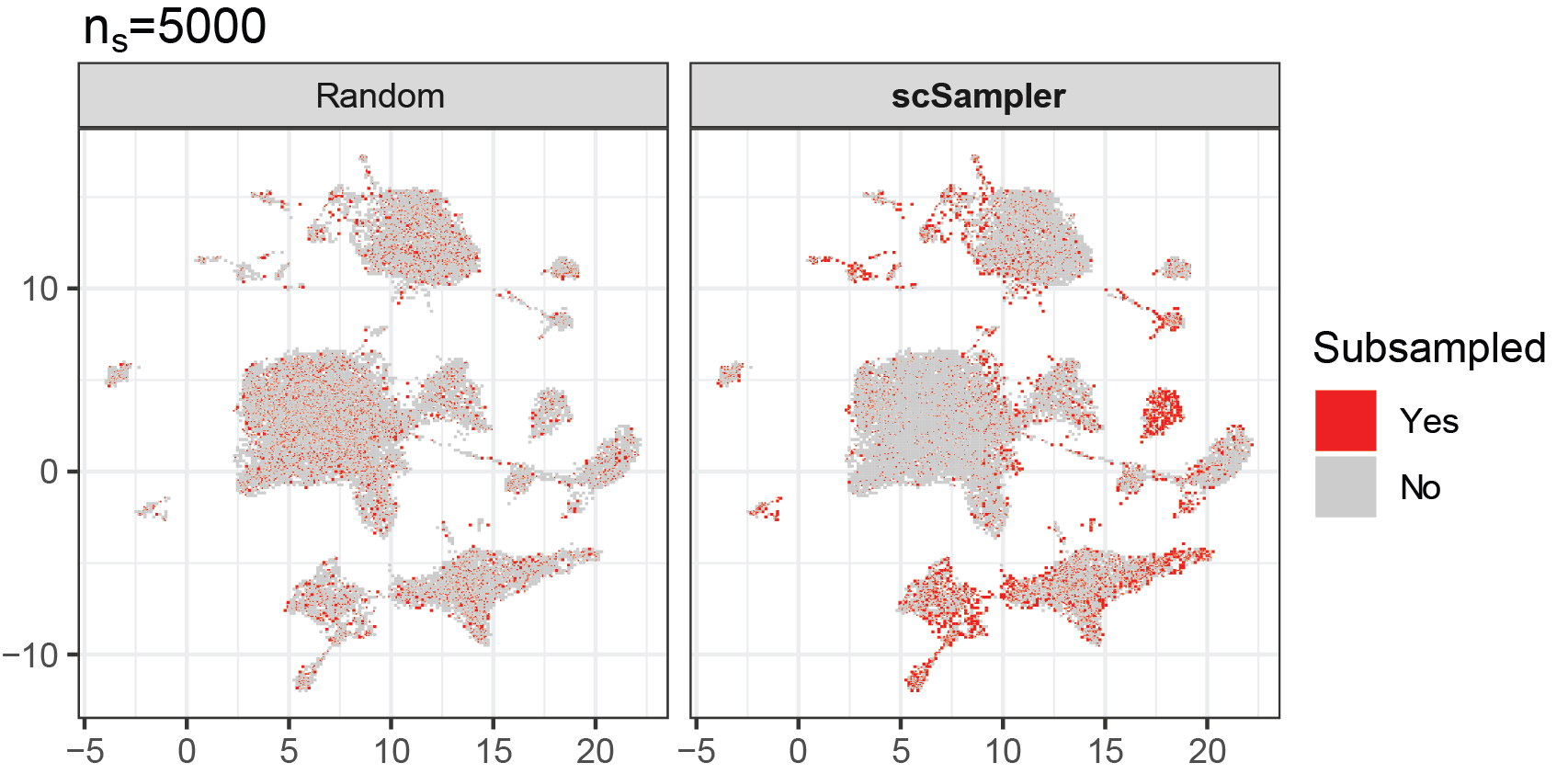
Reducing the complexity of large-scale omics data
How can we reduce the complexity of current datasets with numerous genes and millions of cells? One practical solution is selecting the most informative or representative genes and cells. We developed scPNMF (Bioinformatics; ISMB/ECCB, 2021), a gene selection method that uses dimensionality reduction (Projective NMF) to obtain a sparse encoding of the expression matrix. In addition, we developed scSampler (Bioinformatics, 2022), a diversity-preserving cell subsampling method for large-scale datasets.